5 CAPA best practices for clinical research


Click to enlarge
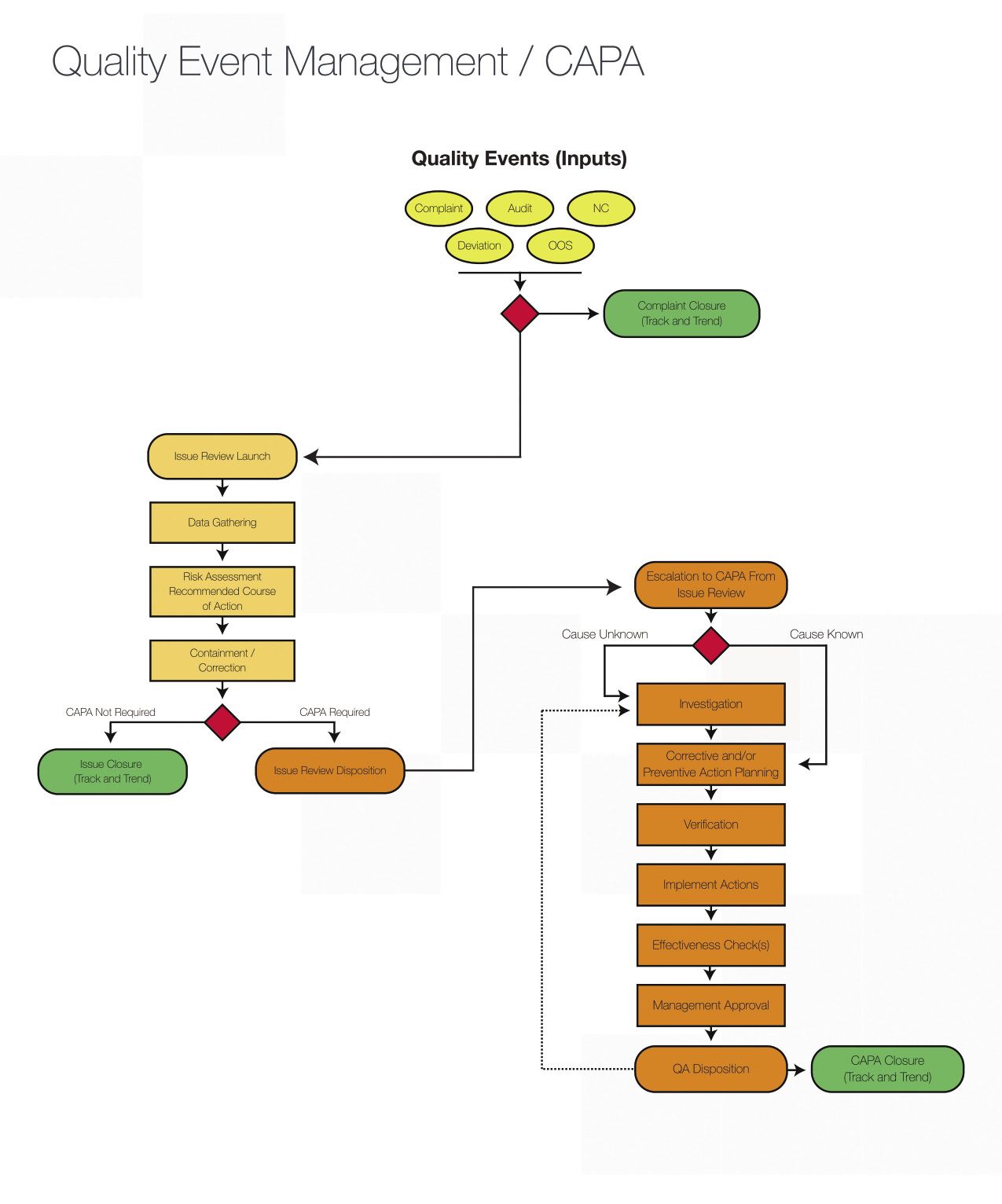
Fig. 1. The diagram shows the CAPA process, from start to finish.[/caption]
In pharmaceutical and biopharmaceutical manufacturing, it is generally assumed that somewhere along the process of manufacturing a product, something is likely to go wrong that could affect quality. When that happens, the quality issue must be resolved in a timely, effective and compliant manner.
If the problem is minor and there’s a solution that can be performed immediately and sufficiently, then the quality event can be closed with an effective containment or correction. If the issue is significant, it will be escalated into a corrective action and preventative action (CAPA).
CAPA is inherently tied to the concept of product quality in manufacturing. Companies regulated by the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and other regulatory entities, as well as companies that adhere to ISO quality standards, ICH quality guidelines, and similar international standards, typically maintain a CAPA process as part of their quality management system (QMS).
So why can’t clinical research professionals adopt CAPA in clinical trials? In fact, they have been using CAPA more than most people realize. But, unlike in manufacturing, there’s less emphasis in formalizing CAPA procedures and embedding them in a clinical trial’s quality process, or in integrating them with the QMS.
Indeed the term “clinical quality management system” or CQMS was coined by MasterControl only five years ago. There is much to be learned from the manufacturing sector when it comes to leveraging CAPA, not only for compliance purposes but to help accelerate time
to market.
CAPA principles
Essential CAPA principles come from regulations and quality standards. For clinical research, the FDA’s Good Clinical Practice (GCP) regulations and the International Conference on Harmonization (ICH) E6 GCP Consolidated Guidance, which FDA has adopted, require a CAPA plan and implementation when quality issues arise. The CAPA should include investigation of how widespread the problems are, correction of the problems, and corresponding efforts to prevent their reoccurrence. [1]
The European Medicines Agency (EMA) Good Practice Guide on Risk Minimisation and Prevention of Medication Errors has similar requirements for clinical trials. It says “when medication errors result in adverse outcomes, corrective actions should be taken.” [2]
CAPA for clinical research
No clinical research is free of quality issues. They are unavoidable, perhaps inevitable, during the life of a clinical trial. What is important is that they are addressed promptly and properly so they will not occur again, and that the scope of the impact of the issue is clearly understood and mitigated.
Given the regulatory requirements and the inevitability of quality problems, most pharma companies have a procedure or a plan for correcting quality issues even if they don’t have a formal CAPA process. At the minimum, most of them have quality-control and quality-assurance processes embedded into their clinical management plans. For some, a separate risk management process is also in place. These three areas work separately but closely together, providing checks and balances in quality management.
If you work in clinical research, you can use CAPA strategies that have been tried and tested in manufacturing and adopt them for your specific needs. The CAPA framework for investigating a quality issue is applicable in clinical as much as in other areas.
Five best practices
Let us take a look at five CAPA best practices that can strengthen clinical research compliance and also help accomplish a fundamental goal across the board in the pharmaceutical industry—accelerating time to market.
1. Establish a CQMS and make CAPA a part of it.
If you are in the process of creating a clinical quality management system (CQMS), integrate a CAPA process from the get-go for a smoother clinical trial. Take advantage of the latest technology and adopt an electronic CQMS for managing CAPA and also your trial master file (TMF). For those using paper-based or hybrid processes, switch to an electronic CQMS to streamline clinical quality data-management through the life of the trial.


Click to enlarge
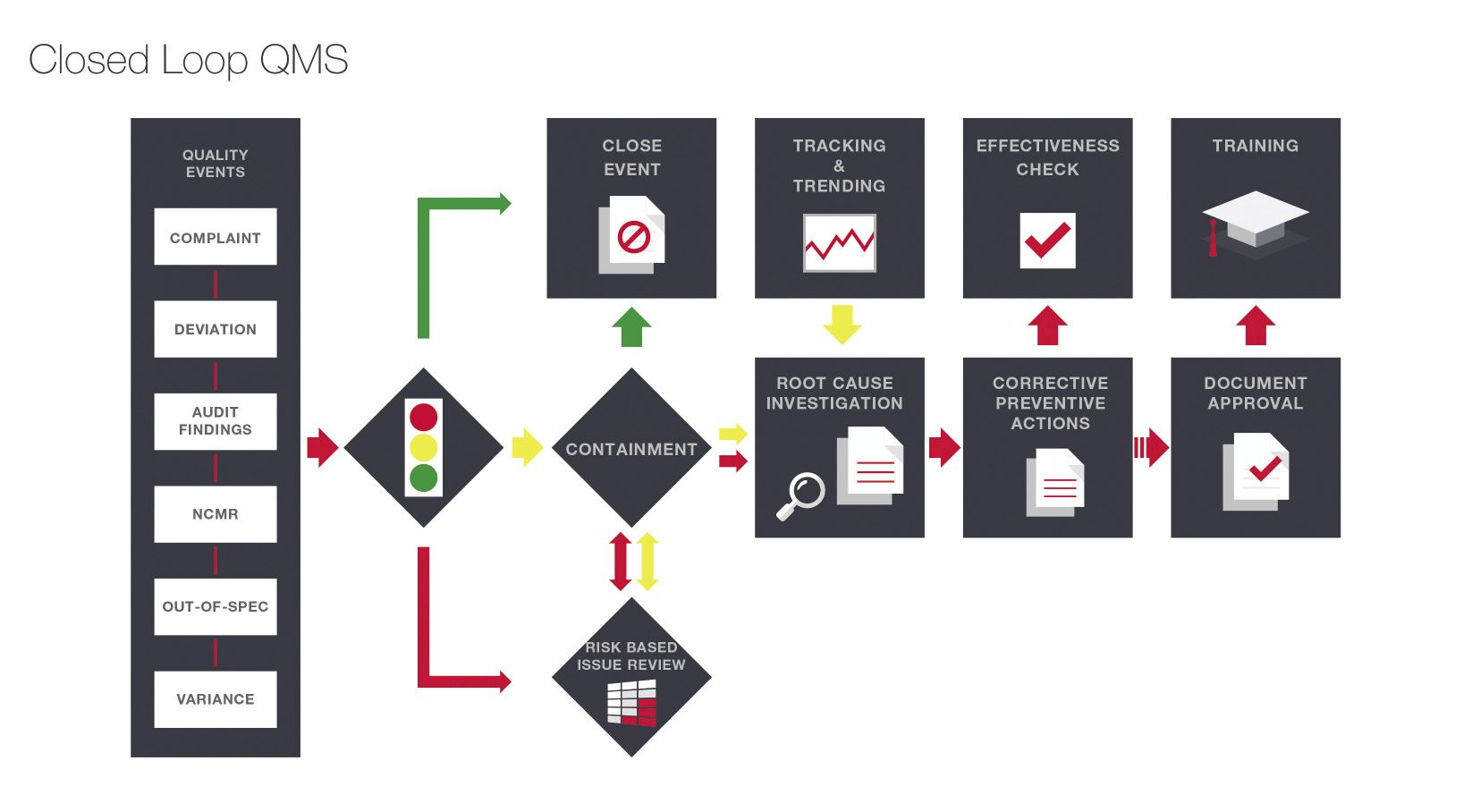
Fig. 2. This is how a robust CQMS integrates a closed-loop CAPA process.[/caption]
Develop a “closed loop” CAPA process as part of your CQMS. This means every quality event must go through various “loops” or stages for an effective resolution, beginning with a determination of the cause of the problem, implementing an appropriate corrective action, making sure the corrective action is working, and closing the loop by eliminating the cause of the problem to prevent it from reoccurring. Choose a CQMS that will guide your users seamlessly from one loop to the next using robust forms and workflow.
A medical device company successfully revamped its CAPA process when it switched to a fully integrated system. The company used to devote every Friday to reviewing and approving CAPAs and other noncompliance issues at a face-to-face meeting. The process was error-prone and wasted a lot of time. The CAPA and training processes were not connected. On top of it all, 10 employees managed documents full time. The company has since switched to a holistic electronic quality system, vastly improving CAPA, training and document management. No more Friday CAPA-marathon meetings. [3]
2. Integrate training control with CAPA to boost compliance.
Clinical trial sites must follow the study protocol and corresponding SOPs to exact specification. One way to make sure that this happens is by training investigators and support staff on the protocol and SOPs, and to conduct ongoing training with regard to protocol amendments.
In the event of a deviation from procedures, it should be documented properly and corrected through the CAPA process. However, corrective actions cannot exist in a vacuum. If a CAPA resulted in a substantial change in a protocol, SOP or some other important document, it will require a retraining of all affected employees. In a paper-based or hybrid process that uses disparate systems, CAPA is not connected to training. Employees could still be performing their jobs based on outdated SOPs, manuals and other documents.
The quality of clinical research results can be significantly affected by inadequate staff training and retraining. An effective training management system will serve as a “gate” that opens when staffers are properly trained and tested, and closes for control (and alerts stakeholders) when training tasks are incomplete or unfulfilled.
3. Integrate CAPA with risk management for a holistic approach to quality.
If your company has a risk management process but not a CAPA process or vice versa, you are not optimizing each process. Use them to complement each other.
Risk management is the task of identifying, measuring and prioritizing the impact of the uncertainty of various variables throughout a clinical trial. For example, a protocol that contains complex tests and procedures has a higher risk of noncompliance than one that contains basic testing only. As part of risk management, you might establish a stricter screening process for selecting study sites. You can integrate such screening protocol in CAPA as a preventative action.
If your risk management and CAPA processes are connected, you will be able to respond in a timely manner. For example, if a site reports a serious deviation from the complex testing during the course of risk assessment, that site will be able to launch a corrective action seamlessly. The staff managing those two processes will be able to communicate and act in unison to resolve the issue.
4. Identify which deviations warrant a CAPA and match the corrective action to the severity of the problem.
Not every quality issue requires a corrective action and not every corrective action calls for a preventive action. The decision on what type of action is needed should be based on a number of factors, such as risk, impact, severity and frequency of the event. The depth of a CAPA investigation and implementation should match the risk. The ICH GCP guidelines acknowledge that not all clinical trial deviations affect the scientific value of clinical trial results. The guidelines outline the types of protocol deviations that should be reported as “serious breaches” and require appropriately serious CAPAs. [4]
A good clinical quality plan should not rely solely on a CAPA process. It should have quality procedures in place from the get-go, before any quality problems occur. The procedures should identify necessary actions prior to, during and after a CAPA is executed. It should also include criteria for evaluating when a CAPA is (or is not) necessary.
An electronic CQMS will equip you with tools for better assessment of quality issues. Choose a system with robust tools, such as CAPA matrix, analytics and reporting tools; best-practice forms for collecting and tracking data for risk assessment; and checklists for monitoring visits.
5. Make your CAPA process transparent to increase your audit and inspection readiness.
All essential documents are subject to audits and inspections. Regulatory agencies evaluate the conduct of a clinical trial and the data it generates by reviewing all essential documents. During an audit or inspection, those documents will be scrutinized.
In a manual process, paper documents are hard to track and can be easily misplaced. There’s nothing worse than failing to produce an important document during an audit. When you automate your CQMS, you will have a single centralized repository for all CAPA documents and other important records. Auditors will need to look at only one place for anything they need. An electronic CQMS makes it easier to search, track and retrieve documents.
For some companies, this best practice can be a hard-earned lesson. A regulated company with presence in six countries adopted an electronic QMS with an integrated CAPA process as a corrective action in itself after an audit. The company used to manage about a hundred CAPAs annually with an ineffective homegrown process that resulted in audit findings. In a complete reversal of fortune, auditors now commend the company’s CAPA process and holistic quality system. [5]
CAPA as a quality tool
Pharma and biopharma companies like yours face the challenge of sustaining growth and innovation in the face of fierce competition in the global market. Noncompliance in clinical research can be extremely costly.
High-risk deviations can result in high numbers of patient dropouts, disqualification of sites, clinical hold, and may even result in the FDA’s “refuse to file” action. Identifying deviations and taking corrective actions in early stages of a clinical trial can reduce cost, reduce risk to patients and increase time to market.
In establishing or implementing a CQMS, take advantage of what the latest technology has to offer to facilitate compliance with GCP requirements throughout a clinical trial.
References
- FDA GCP/Clinical Trial Guidance Documents, from FDA website http://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/GuidancesInformationSheetsandNotices/ucm219433.htm
- MA’s Good Practice Guide on Risk Minimisation and Prevention of Medication Errors, from the EMA website, http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2015/11/WC500196981.pdf
- A Case Study from the Perspective of Executives: MasterControl Helps MicroMed Leverage Resources, published by MasterControl, 2013.
- E6 GCP Consolidated Guidance, from the ICH website: http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/good-clinical-practice.html
- In 2009, Chart Industries successfully overhauled its quality system and improved its CAPA process by switching from a paper-based QMS to MasterControl All Access, which integrates CAPA, training, audit, change control and other modules.
ABOUT THE AUTHOR



http://www.mastercontrol.comPatricia Santos-Serrao, RAC, is the product management director of clinical and regulatory solutions for life sciences at MasterControl. She has two decades of experience in regulatory affairs and clinical areas in the pharmaceutical industry, including working for Schering-Plough and Boehringer Ingelheim. She earned her Regulatory Affairs Certification from the Regulatory Affairs Professional Society and the Regulatory Affairs Certification Board.
Save
Save
Save
Save




Newsletter
Stay ahead in the life sciences industry with Pharmaceutical Commerce, the latest news, trends, and strategies in drug distribution, commercialization, and market access.











Multi-Indication Drug Branding in Today’s Pharmaceutical Industry
July 2nd 2025As drug development increasingly targets multiple indications, pharma companies must make strategic branding decisions—balancing regulatory requirements, market dynamics, and patient safety—to choose between single-brand or multi-brand approaches.






