The IRA is Industry's Wake-up Call
Exploring the legislation's diverging requirements between regulatory agencies and global payers, and its impact on the commercial results of biopharma companies
The recently approved Inflation Reduction Act (IRA) of 20221 is highlighting an issue that the biopharmaceutical industry has failed to address and that can no longer be ignored. Now that the US is effectively introducing price controls for at least part of the prescription drug market, it is time to consider payer and provider needs more seriously in drug development planning. The IRA comes at a time when outside the US, the biopharma industry is increasingly faced with payer rejections of new drug value propositions and subsequent market withdrawals or poor commercial results. Bluebird Bio’s withdrawal of Zynteglo from Europe after failed price negotiations was just one example of misalignment between biopharma expectations and commercial reality.
Inadequately addressing payer evidence needs is already leading to suboptimal pricing, and sometimes, an inability to launch in international markets. It is time that the industry realizes that placebo-controlled clinical trials, single arm trials, and surrogate endpoints may pass the FDA hurdle but may not lead to commercial viability. Biopharma companies now need to address a lingering value issue that they have so far largely ignored.
CMS 'negotiations'
The IRA includes a redesign of Medicare Part D, allows the Centers for Medicare and Medicaid Services (CMS) to negotiate drug prices for Medicare Parts B and D, and penalizes inflationary price increases with Medicare rebates. The most concerning part about the new law is the “negotiation” aspect. The level of penalties for not complying with CMS rules gives a strong scent of direct price control rather than negotiations; this is also likely a basis for legal challenge of the IRA.
We must wait for further details on the implementation of the CMS negotiation framework. The IRA seems to have a relatively favorable impact on biologics vs. small molecules and on pharmacy benefits vs. medical benefits, although it remains to be seen how managed care will adjust its formulary management after patient cost for Medicare Part D is capped at $2,000 per year.
Assuming that (after legal challenge or not) negotiations will be based on value and competition, there is a high probability that the steepest discounts will fall on drugs that are medically deemed undifferentiated or insufficiently substantiated with evidence of patient or healthcare system value.
Ex-US payer requirements
In many countries outside the US, drug price controls have dominated the commercial landscape for decades. Increasingly strict clinical and economic evidence requirements create steep commercial hurdles beyond the efficacy and safety-oriented regulatory approvals, such as by the European Medicines Agency (EMA) that officially signify market authorization, but in reality, just start payer discussions.

Amplified by COVID-19-related general economic and healthcare budget pressures, global governments are engaging in cost-cutting programs that include strict containment of prescription drug spending. ZS analysis2 shows that ongoing healthcare budget deficits will likely lead to more severe restrictions and lower prices or price cuts for undifferentiated drugs, increased scrutiny on value of high-budget impact therapies, increased use of biosimilars, and price pressures on combination oncology treatments and orphan drugs. As an illustration, the share of reimbursement submissions in France that were successful in securing a rating of ASMR I, II, or III, thus having a potential for a meaningful price premium over the standard of care, has declined from only 7.1% from 2006-2010 to an even lower 3.6% from 2016-2020. In Germany in 2021 and the first half of 2022, nine new drugs were withdrawn from the German market over health technology assessment (HTA) results and resulting pricing problems. Prior to 2020 this was a very rare event.
Payers increasingly scrutinize a lack of longer-term outcomes as proof of drug benefit. Where the medical community and FDA or EMA tend to accept surrogate endpoints as evidence of efficacy, payers demand evidence of sustained effectiveness. For example, payers look for a meaningful improvement in overall survival rather than a tumor response rate, or they demand a reduction in cardiovascular event rates instead of lowered cholesterol. Biopharma companies often do not take these requirements seriously, sometimes because they mistakenly believe these hurdles can be overcome and other times because they are simply not willing to take the clinical risk or make the additional time and monetary investment to show compelling data.
Typical evidence gaps
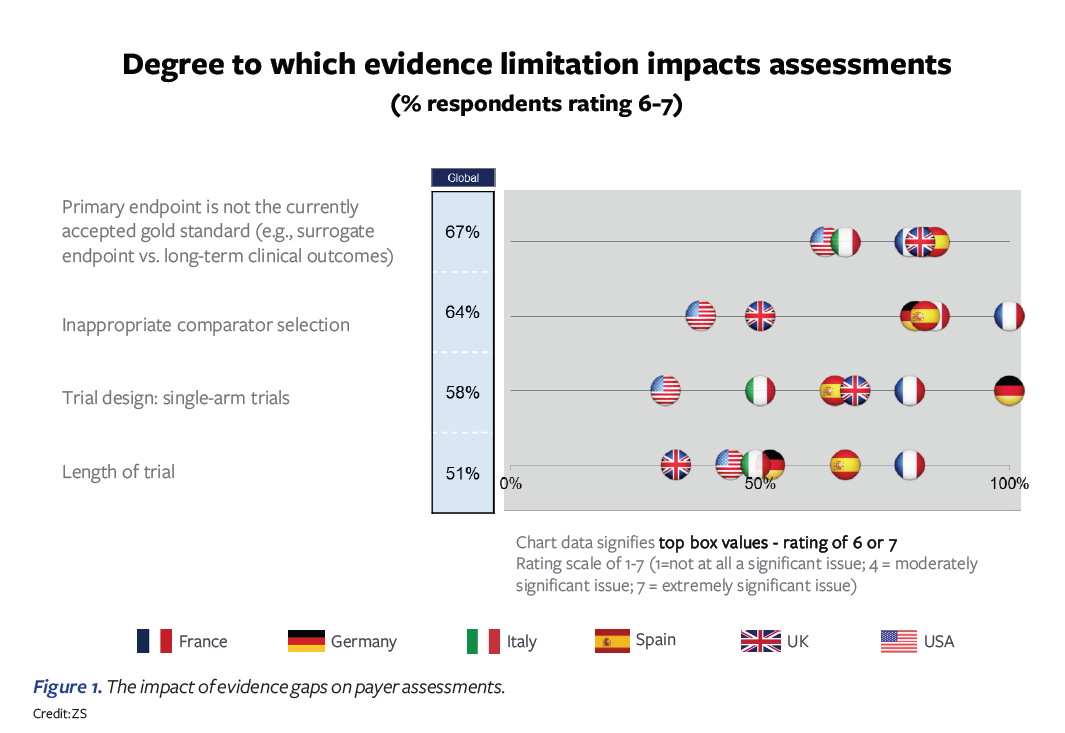
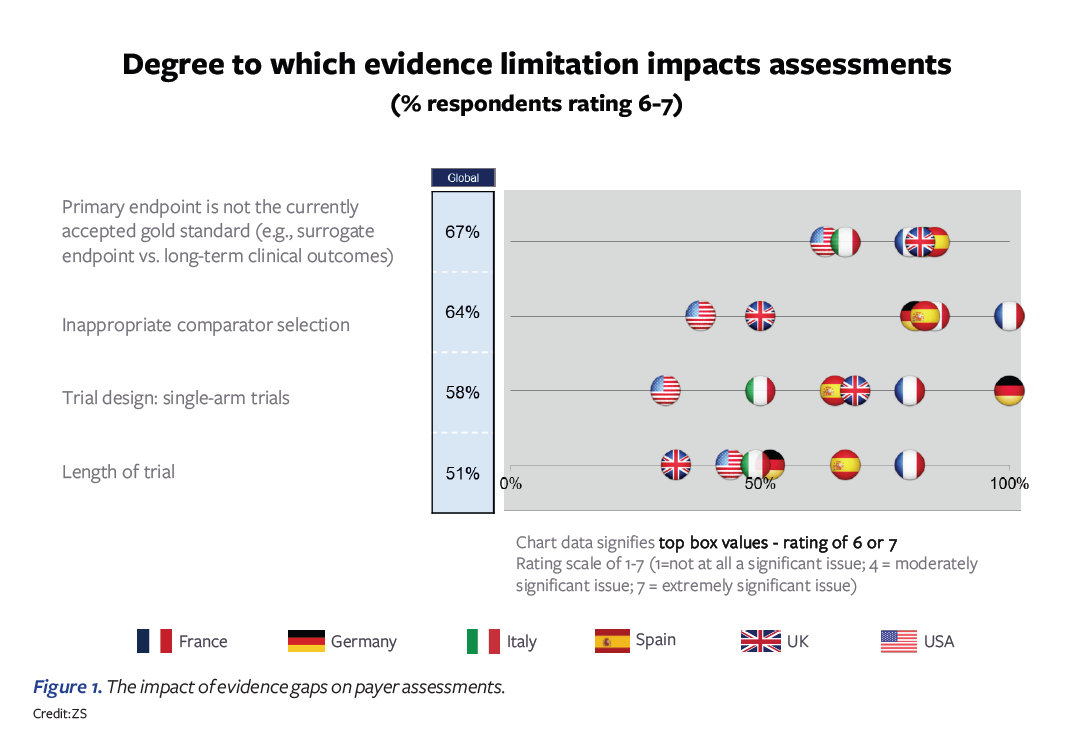
In an early 2022 poll, we asked payers in the US, Germany, France, Italy, Spain, and the UK to give their perspectives on biopharma pricing and reimbursement submissions and the main gaps that they experience. Four evidence gaps were highlighted as having the largest impact on payer assessments:
- Primary endpoint not accepted by payers
- Trial comparator is deemed inappropriate
- Single-arm trials
- Length of trial insufficient
Figure 1 above illustrates that the ratings across countries vary somewhat. Overall, France and Germany tend to be most critical in their evidence requirements; ironically, these are also the most-referenced countries among the set, making pricing and access success in these two countries critical for ex-US global commercialization. In France, the choice of trial comparator is the highest-ranked concern, while in Germany, the single-arm trial is highlighted most. Spain and Italy also express substantial concerns. The US and UK are on the lower end of the concern spectrum and tend to be more willing to find ways of overcoming the evidence gaps through other analyses. For the US, the question is how this may change post-IRA.
Primary endpoint not accepted by payers
The selection of the primary endpoint in pivotal clinical trials is the most frequently stated payer concern expressed across countries. US payers also express strong concern about this aspect. It reflects the consistent feedback from payers that, while clinicians make decisions based on surrogate endpoints as immediate signals on patient management, payers are highly skeptical whether, for example, in oncology, tumor response rates, or even progression-free survival (PFS) are leading to meaningful survival improvements. As a result, drugs with lack of compelling evidence of long-term patient health improvements are heavily penalized on price in countries such as France and Germany. An example is the CDK4/6 class, where Ibrance, Verzenio, and Kisqali received relatively poor pricing outside the US despite compelling PFS data, as these therapies lacked evidence of overall survival improvement at launch.
Trial comparator is deemed inappropriate
The choice of a comparative treatment arm in a randomized controlled head-to-head clinical trial is critical in many payer systems. Having chosen an inappropriate or no-longer-relevant comparator can have a drastic impact on the accepted benefit level. An example of an unfortunate outcome has been the withdrawal of Piqray3 from Germany and France after payers deemed the comparator treatment arm in the trial no longer the relevant choice since the adoption of CDK4/6 inhibitors. Particularly in France, Germany, Spain, and Italy, this is one of the most common reasons for an unsuccessful price negotiation.
When an appropriate comparator is not selected, payer acceptance comes down to the justification. It may not be a surprise that, what is often the real reason (“I am not sure I can win against product X”) is exactly the reason why payers insist on this point. Payers may say, “If you don’t have the stomach to show value, why do you expect us to pay for it?”
Single-arm trials
Single-arm studies are only deemed appropriate in situations where a properly powered head-to-head trial showing statistically significant improvements on important endpoints is not feasible, such as in the situation of many orphan drugs. Yet even in this situation, payers have become very critical. Germany is increasingly rejecting benefit claims for orphan drugs over lack of evidence through the single-arm trial, which we saw clearly illustrated by the German responses in our survey. This should be seen in the context of the German government’s actions to clamp down on what it considers an explosion of orphan-drug applications following Arzneimittelmarkt-Neuordnungsgesetz's (AMNOG) automatic exclusion of “no-benefit” designations for orphan drugs with limited revenue.
Length of trial insufficient
Payers are also concerned with a sustained effect of treatment. Shorter trials can satisfy claims regarding some surrogate endpoints. But initially, observed improvements can be reversed over time, resulting in a lack of longer-term improvements in outcomes. There is some overlap between this concern and the insistence on endpoints that reflect longer-term outcomes. For example, in kidney disease, a sustained change in estimated glomerular filtration rate (eGFR) may support claims of disease modification, although payers usually want to see a verification in terms of fewer patients progressing to end-stage renal disease.

Real-world data to the rescue?
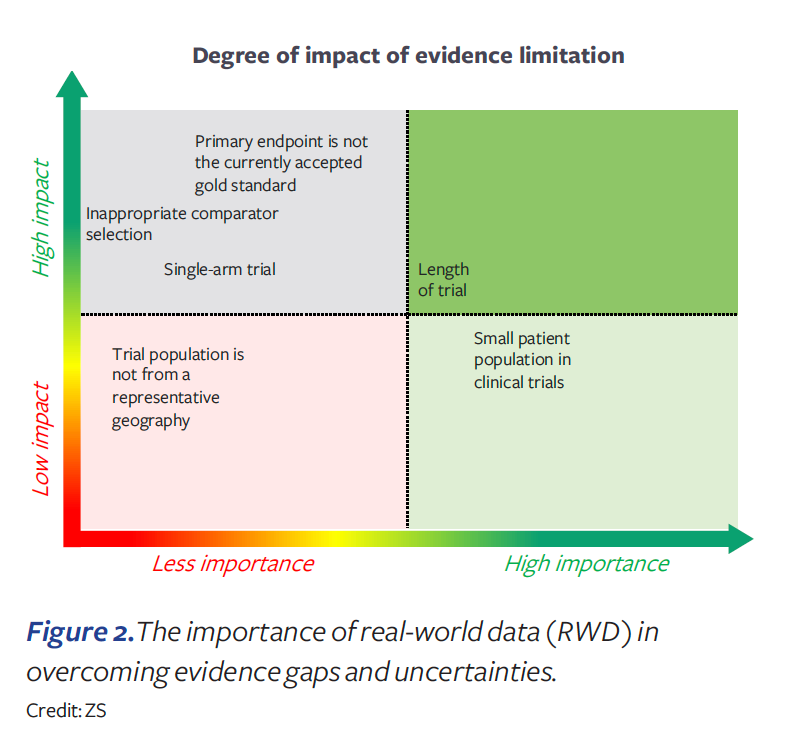
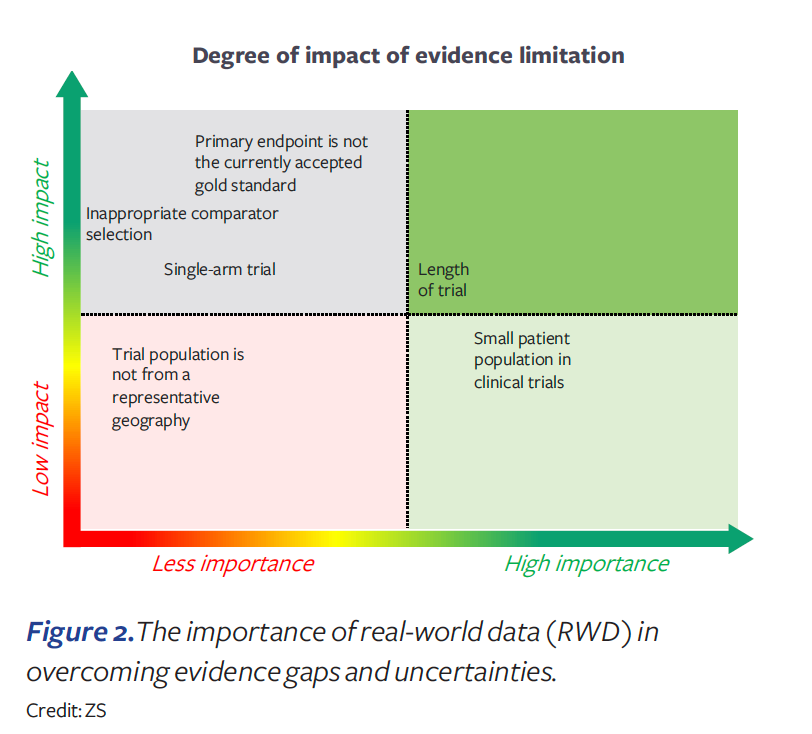
The medical community has great expectations for real-world data (RWD). RWD offers opportunities to learn from statistical analyses of real-life outcomes across patient populations and treatments. But RWD can only be obtained in support of benefit claims after widespread use in medical practice and thus has limited use for initial approvals for new drugs. Since the 21st Century Cures Act of 2016, FDA has become more engaged in addressing the role of RWD in US marketing authorization approvals. Recent examples4 show some FDA willingness to use RWD as an external control arm and in addressing concerns regarding single-arm studies. However, payers are only slowly and highly selectively accepting RWD.
In our survey, payers strongly indicated that while RWD can be useful for treatment protocols and to orient physician prescribing, they do not accept uncontrolled observational data as a substitute for randomized controlled trial data. More specifically, payers indicated that RWD will generally not be accepted to overcome an inappropriate endpoint selection or trial comparator, or for choosing to conduct a single-arm trial (see Figure 2). Therefore, we should be very careful in assuming that evidence gaps can be fixed with RWD.
Three imminent actions for biopharma
Many biopharma companies are still coming to grips with the need to demonstrate evidence of value beyond the very limited requirements that FDA is imposing. This is often a deep organizational issue, where development decision-making is heavily focused on speed and clinical risk reduction rather than a holistic evaluation of multiple options with respect to impact on commercialization. Many companies make market share projections based on theoretical potential, but ignore evidence needs to ensure that the potential comes to fruition. They tend to choose the fastest road to FDA approval, largely ignoring the evidence needs of the medical community, payers, providers, and prescribers. As a result, they lack acceptance in guidelines and have poor formulary placement and slow adoption.
There are several things that biopharma companies can do today to be better prepared for the evolving needs of payers and the emerging needs under the IRA.
- Enforce better-structured access involvement in the drug development and corporate business development decision-making processes. The creation of a chief access officer is long overdue to ensure that access has a strong voice next to R&D, to add a sense of urgency, to build a stronger organizational understanding of payers and providers, and to pursue meaningful customer partnerships.
- Incorporate access and pricing in forecasting and business decision-making. Medical community adoption in guidelines, payer/provider formulary uptake, and physician prescribing are highly correlated with the evidence provided beyond the FDA-mandated placebo-controlled trial. Forecasting needs to support trade-off analyses and decision-making that considers multiple development options. Scenario planning is important to address uncertainties with respect to further US market evolution within and beyond the IRA implementation.
- Evolve global strategies to address commercial evidence needs in the US, Japan, Europe, and emerging markets more explicitly. Consider regional evidence needs, and not just as a US afterthought. In some cases, it may make sense to launch in the US and Japan first, and strengthen US market uptake and European/Canadian pricing and access requirements with more robust evidence. However, price control timelines under the IRA could change that.
About the Author
Ed Schoonveld is a managing partner and founder of the value and access practice at ZS, a management consulting and technology firm.
References
1. https://www.zs.com/insights/inflation-reduction-act-what-biopharma-needs-to-know
2. Stefan Kloss and Emmanuel Colliot, “Once bent out of shape: Implications from the latest cost containment measures to address post-COVID healthcare budget deficits in Western Europe,” ZS white paper, Oct. 2022.
3. Neena Brizmohun, “Piqray: German Withdrawal, European Reimbursement Deals & Broader Indication Plans,” Pink Sheet, May 6, 2022. https://pink.pharmaintelligence.informa.com/PS144276/Piqray-German-Withdrawal-European-Reimbursement-Deals--Broader-Indication-Plans#:~:text=Novartis%20has%20withdrawn%20Piqray%20(alpelisib,use%20across%20Europe%20last%20year
4. Bridget Silverman. “RWE On The Runway: Trio Of Upcoming Approval Decisions Will Test US FDA’s Thinking On RWE For Efficacy,” Pink Sheet, Sept. 5, 2022. https://pink.pharmaintelligence.informa.com/PS146958/RWE-On-The-Runway-Trio-Of-Upcoming-Approval-Decisions-Will-Test-US-FDAs-Thinking-On-RWE-For-Efficacy

")

























