Biomarkers drive advances in drug therapy
As the definition of ‘targeted’ therapies—especially in oncology—and as a component of companion diagnostics, biomarkers are creating opportunities in drug research and commercialization
In the abstract, a biomarker is any quantifiable aspect of a living system that indicates its relative health; an old definition from the World Health Organization characterized it as “any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease.” In today’s pharma industry, however, biomarkers have taken on more specific meanings: measurable features that characterize the presence of a disease or condition in patients, especially immunological features, and most recently, information on the genetic profile of patients or their disease (such as the genetics of a tumor, which are distinct from the genetics of the cancer patient).
In practice, biomarkers are affecting—in fact, enabling—the development of new drugs, either as a way to identify the origin of some genetic conditions or birth defects, or as a way to identify which patients will respond to a particular therapy. For some approved, commercial drugs, biomarkers are the heart of “companion diagnostics”—tests or measurements that show alignment between a particular patient’s disease, and a particular therapy. Over the past 20 years, FDA has provided multiple guidances on both biomarkers as clinical endpoints, and biomarkers as companion diagnostics.
While biomarkers and companion diagnostics are relevant to a wide range of diseases or genetic conditions, most of the focus today is on biomarkers for treating cancer. To date, thousands of biomarkers, across 18 different therapeutic areas, covering nearly 2,900 therapeutic indications, have been identified, with detailed information centralized in the comprehensive Global Online Biomarker Database (GOBIOM) that is maintained by Excelra Knowledge Solutions Pvt. The National Cancer Institute maintains a similar database, an activity of the Early Detection Research Network.
In general, companion diagnostic tests (CDx) that are used to identify targeted genetic mutations and other biomarkers reach the market in two ways:
- In vitro diagnostic (IVD) kits are developed by specialized diagnostics companies, reviewed by FDA (or other regulatory agencies) and distributed to certified laboratories to allow for decentralized testing for large patient populations.
- Laboratory-developed tests (LDTs) are developed and validated analytically at a local laboratory (such as an onsite lab operated at a major oncology hospital or research center) and then used for centralized testing at that location.
FDA has a “List of Cleared or Approved Companion Diagnostic Devices” on its website; as of January, 36 tests are listed there; all but two are for various cancers, and all are tied to one or more drug therapies. Additionally, there are over 120 “nucleic acid-based” (DNA) tests, and the great majority of these are also directed at cancers. The second category—LDTs—has been the subject of repeated examinations by FDA, but to date no action has been taken on a premarketing or pre-certification program by the agency. A “discussion draft” of a bill (the VALID Act) was introduced to Congress in December.
Market analyst Kalorama estimates the size of the 2018 US precision cancer-testing market at $8.1 billion, split roughly evenly between IVD testing and imaging. Another market research firm, QYResearch, Inc., recently estimated the North American LDT market at $2 billion.
A steep learning curve, shared by all
“All of today’s oncology agents have some level of toxicity or adverse events that may necessitate emergency room visits and hospitalizations. It doesn’t make sense to give patients toxic therapies — which can cost tens of thousands of dollars per month, and produce no meaningful clinical benefit — when screening for the targeted biomarker can help providers to match the patient with the best therapy right from the outset,” says Roger Brito, DO, National Director of Oncology for Aetna. “The rise in targeted oncology agents has truly become a game changer, and payers tend to look very favorably upon it. The more evidence there is to attach value by showing that the therapy is delivering quality care, the better.”
The Excelra GOBIOM database, with its thousands of therapeutic indications, lists fewer than three dozen genomic biomarkers that are currently included in the label indications of existing FDA-approved therapies, including:
- BRAF mutation in melanoma, thyroid, colon and lung cancers
- KRAS mutation in colorectal and lung cancers
- HER2+ amplification in breast cancer
- ALK+ mutation in non-small-cell lung cancer, lymphoma, neuroblastomas and other
- EGFR mutation in non-small-cell lung cancer (NSCLC)
- BRCA mutation in breast, ovarian, prostate and pancreatic cancers
- Microsatellite instability-high (MSI-H) and mismatch repair deficient (dMMR) mutation in colorectal cancer and more
- NTRK mutation in NSCLC, thyroid, colon, pancreatic, breast cancers and more.


Last November, Roche introduced the Ventana pan-TRK Assay for detecting TRK protein fusions. Depicted (left): salivary gland carcinoma; (rigth) squamous cell carcinoma. The Roche test stains the fusion proteins in multiple types of tumors. credit: Roche
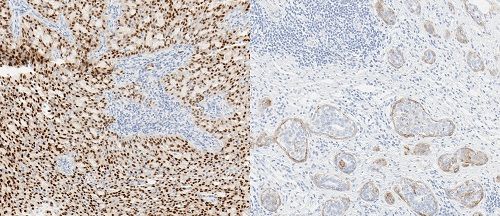
According to FDA, progress in biomarker qualification is being stymied by a lack of a clear and predictable and specific regulatory framework for their use. To rectify this, FDA’s Center for Drug Evaluation and Research issued its Biomarker Qualification Draft Guidance in December. The proposed framework recommends key components of a biomarker-development program to systematize both the type and level of evidence necessary for biomarker qualification, drug development and clinical trial design.
The sweet spot is often elusive
“When Genentech’s Herceptin (trastuzumab) was first commercialized, there was an opportunity to think this would be a good therapy for anybody with metastatic breast cancer, but scientists soon realized it would work better with patients with metastatic breast cancer who overexpress the HER+2 estrogen receptor (human epidermal growth factor receptor 2),” says Nicholas Robert, MD, Medical Director, Data, Evidence & Insights for McKesson Life Sciences and the US Oncology Network. “This concept led to the successful use in patients with early-stage disease. If Genentech had not made and validated this discovery early on, it would have missed the chance to show how successful this drug can be. Instead, Herceptin has turned out to be an absolute winner — and a market blockbuster — by directing its use to just a subset of about 20% of all patients whose metastatic breast cancer has demonstrated the HER+2 mutation.”
By contrast, Robert notes that Genentech also developed Avastin (bevacizumab), a tumor-starving anti-angiogenic agent, “but has been unable to find biomarkers to reliably identify which patients will benefit from it.” Avastin is approved for use to treat certain types of colorectal, lung, kidney, cervical and ovarian cancer and glioblastoma (and even wet age-related macular degeneration). “However, some patients respond well while others do not, and the inability to identify a reliable biomarker to help guide its use more precisely leads to a lack of specificity that has really limited the drug.”
Another industry observer notes that AstraZeneca “made a tactical error” when it first introduced Iressa (gefitinib). When it was first approved in 2003, Iressa, a kinase inhibitor, was being to treat patients with non-small cell lung cancer (NSCLC), but FDA later withdrew its approval in 2005 due to lack of evidence demonstrating it extended life meaningfully. “This happened just at the point when the epidermal growth factor receptor (EGFR) mutation story was starting to emerge, and initially AstraZeneca missed better opportunities to focus its drug on patients whose NSCLC had the EGFR mutation.” Today, Iressa, has experienced a comeback and is now approved for use in patient with certain breast, lung and other cancers that test positive for mutated and overactive EGFR.
While the fragmentation of patients into go/no-go cohorts does diminish the ultimate market potential for the drug, “there’s still general agreement that even smaller, niche patient populations can still be very valuable for drug makers, and the ability to demonstrate stronger clinical outcomes in a biomarker-directed patient population is always a good business strategy,” says Brad Smith, PhD, Vice President of Translational Medicine for IQVIA. “In oncology, the ability to target unmet need has been shown to accelerate the drug-approval process and drive immediate market uptake.”
“It’s always preferable to get the investigational therapy approved — even in a narrower indication that has more strong responders — and then work to expand the number of approved indications over time,” adds Nicholas Dracopoli, PhD, SVP, Personal Genome Diagnostics (PGDx). “This allows the research community to then build on that first approval over time, for other uses, other indication and other combinations.”
“By reducing the use of older cytotoxic therapies that may bring significant toxicity or side effects, and seeing longer survival periods for patients, a picture begins to emerge of the total value, and this can help payers to justify coverage of a premium drug price and the cost of diagnostic testing,” adds Susan Weidner, SVP, IntrinsiQ Specialty Solutions, a unit of AmerisourceBergen.
Biomarker breakthroughs enable ‘tissue-agnostic’ therapies
One of the biggest advantages of biomarker-directed therapies is the ability to identify certain actionable genetic mutations that occur in malignancies across many different organ systems (for instance, lung, breast, colorectal and more) but have the same functional effect in each — an important phenomenon that has opened the door for so-called “tissue-agnostic” therapy options. Any oncology therapy that is approved for use in multiple types of cancer helps the lifesaving drug to reach more patients, and helps the drugmaker to reach a larger potential market size.
To date, two tissue-agnostic oncology therapies have been approved by FDA:
* Keytruda (pembrolizumab), a checkpoint inhibitor immunotherapy agent from Merck, was originall approved by FDA in 2014 for melanoma. In 2017, its indication was expanded for numerous other cancers that shared MSI-H or dMMR mutations; the first such approval from FDA.
* Viktravki (larotrectinib) from Bayer and Loxo Oncology, Inc., approved in November 2018. It was approved without a specific, designated companion diagnostic (CDx) test, but targets cancers that harbor NTRK (neurotrophic tropmyosin receptor kinase) fusions. This initial indication was approved under accelerated approval, based on trial results involving just 55 patients. Because of this small patient cohort, FDA says that continued approval may be contingent upon verification of clinical outcomes in further confirmatory trials.
“Vitrakvi targets NTRK fusions that are found in diverse cancer histologies — including both rare cancers with high TRK fusion frequency, and common cancers with low TRK fusion frequency — and there are roughly an estimated 1,500–5,000 patients harboring TRK fusion-positive cancers in the US annually,” says Arturo Loaiza-Bonilla, MD, MSEd, FACP, Vice Chairman of the Dept. of Medical Oncology, Cancer Treatment Centers of America (CTCA), and Chief Medical Advisor and co-founder of MassiveBio. “You may consider with such a low number of patients that no one would even consider developing this drug. However in the precision oncology space, having a tumor driven by TRK is like hitting the lottery. The overall response rate (ORR) with Vitrakvi was 75%, and response duration was 6 months or longer for 73%, 9 months or longer for 63%, and 12 months or longer for 39% of patients.”
Importantly, Loaiza-Bonilla notes that these numbers can be translated into compelling health economics and outcomes data, which may allow payers to justify coverage for these drugs. For example, if the pharmaceutical company can analyze real-world data (RWD) and ‘synthetic’ control arms using historical patient data from prior trials to compare how those patients would have done otherwise, the results can certainly help to justify the cost of these advanced therapies. “In the case of NTRK fusion drugs, such studies can show that without these inhibitors, patients would have succumbed to the disease significantly earlier, or even required costly hospitalizations and less effective standard regimens.”
“Keytruda’s approval for MSI-H tumors also serves as a proof-of-concept for tissue-agnostic therapy development,” says Jillian Scaife, a principal at Trinity Partners, while noting that there are at least 37 so-called “basket trials” (studying 31 unique therapies) currently being conducted, to evaluate how each targeted therapy performs in tissue-agnostic indications. bring trialed in various mutation- or biomarker-defined, tissue-agnostic indications. When the underlying science supports it, “this development approach could provide access to the greatest number of patients with the biomarker/mutation, and a fast route to market for drug developers,” she adds.
The Targeted Agent and Profiling Utilization Registry (TAPUR) Study, [1] led by the American Society of Clinical Oncology (ASCO), is one multi-center basket clinical trial being carried out at the Cancer Treatment Centers of America. Its goal is to assess how 17 targeted oncology therapies that are already in use, from seven pharmaceutical companies, function across a range of cancer types that share the same genetic mutation — but for which there is not yet an approved label indication for use. When such oncology agents are used off-label, they are often not covered by insurance companies — putting the therapy option out of reach for patients and physicians. If the study pans out, the label indications of the drugs might be expanded.
Creating a rational diagnostic testing rationale
A fundamental question, still being hotly debated, is what level of diagnostic testing is appropriate across the entire landscape of patients who receive cancer diagnosis? Is it cost-effective to carry out potentially costly diagnostic testing thousands of patients, hoping to find a biomarker that may occur in only, say, only 1-in-1,000 patients? Some say that type of population-wide screening is not justified, while others argue that broader diagnostic testing can help payers to reduce their exposure to unnecessary drug costs, and can reduce the patient’s exposure to costly, potentially toxic therapies that promise no clinical benefit. To date, no consistent payer framework has yet emerged, so coverage decisions still tend to vary from payer to payer, and from test to test.
“We may some day get to universal testing for all patients, to identify all actionable genetic mutations, but we’re not there yet. It doesn’t make sense, from a practical standpoint, to carry out very broad panel testing for all patients — with high failure rates and high costs — just to coax out a needle in a haystack with that one patient who may have the rare target mutation,” says Smith of IQVIA. “The scientific community needs to continue pursuing ways to carry out the testing more efficiently.”
“Payers are now looking more deeply into whom to cover for diagnostic testing, and recent FDA and CMS approval of some of these tests are driving the tide of change,” adds Loaiza-Bonilla of CTCA. “Most of the objections are related to the timing of the test being performed, or when it is repeated without justification. The real justification should be focused on finding the right biomarkers to approve therapies and for clinical trial enrollment.”
A recent Cardinal Health study shows that “Even when there is compelling clinical evidence for testing, 25% or more of physicians do not order the test,” says Bruce Feinberg, DO, Vice President and Chief Medical Officer for Cardinal Health Specialty Solutions. And in cases when physicians do test, some still rely on their own judgment, rather than test results, to drive prescription decisions. “Until payers see evidence that testing is being used with consistency to drive improved outcomes and lower costs, they may remain reluctant to cover these costs.”
“The same issue is arising today with the widespread availability of broad, ‘panel-type’ genetic testing,” says Brito of Aetna. “Should we be testing every person for every genetic mutation — even those that are not yet actionable? Such open-ended results have repercussions.” He adds: “Insurers will cover an actionable mutation all day long, but when we’re asked to take on a ‘let’s test everybody for every mutation’ policy, or cover the testing of 400 mutations, that may not have any action, that’s a little bit irresponsible and creates a lot of anxiety in an already anxious therapeutic space.”
“Medicare is fairly consistent in how it evaluates diagnostic testing requirements as a prerequisite for specific treatment,” adds Melissa Elder, Market Analyst for Kalorama. “Since most other third-party payers tend to follow Medicare coverage guidelines, it is important for drug and companion diagnostic test manufacturers to focus on grabbing CMS’s attention with a strong story.”
Drug and test partnering
“Drug manufacturers often want to partner with one diagnostic testing company, which can develop a viable companion diagnostic test, assist with clinical trials, manage the data and seek FDA approval, but they also want flexibility in testing once the drug has entered the market, so that prescribers are not tied to one test provider to do pre-screening, but instead have access to different testing laboratory options to reduce costs, once the drug has entered the market,” says Smith of IQVIA. These days, it is common for the label indication not necessarily to specify one test or lab, but to allow providers to broadly use “any FDA-approved test” to screen patients for the target genetic mutation (such as EFGR or BRCA or other).
“The story may be a little different for recently approved Vitrakvi as the NTRK gene fusion can only be detected by sensitive and specific tests like NGS and FISH,” says Scaife of Trinity Partners. “Loxo Oncology has provided more specific guidance on commercial laboratories that offer testing that can identify the NTRK gene fusion.” TRK-fusion cancer can be detected through a number of diagnostic tests, but only sensitive and specific tests can reliably detect NTRK gene fusions.
According to Loxo, the commercial laboratories that currently offering testing that include NTRK1, NTRK2 and NTRK3 include Foundation Medicine, Caris Life Sciences, Cancer Genetics Inc., Sirona DX, PathGroup, Neogenomics, Knight Diagnostic Laboratories, OmniSeq, Paradigm and Tempus. [2]
In late November, Roche launched its Ventana pan-TRK (EPR17341) immunohistochemistry assay, which is said to be the first automated in vitro diagnostic (IVD) immunohistochemistry (IHC) assay that can detect TRK proteins across many solid tumor types, including lung, thyroid and sarcoma, infantile fibrosarcoma, secretory and juvenile breast cancer, where TRK fusion proteins are likely to be the defining genetic feature, according to the company.
Many in the oncology space heralded FDA’s approval in late 2017 (and the first commercial availability in March 2018) of Foundation Medicine’s FoundationOne CDx as a watershed event that will likely usher in a new era in diagnostic testing. FoundationOne CDx is the first comprehensive diagnostic test (based on next-generation sequencing; NGS) to provide broad genomic profiling of solid tumors across a range of cancers. According to the company, the ability to integrate this comprehensive, single-site assay diagnostic test into clinical care is expected to eventually help one in three patients, across five common advanced cancers, to match more appropriately with FDA-approved biomarker-directed oncology therapies (including immunotherapies), and to help physicians identify patients who meet targeted clinical trial criteria.
According to Foundation Medicine, the FoundationOne CDx diagnostic test is able to assess all classes of genomic alterations in 324 genes that are known to drive cancer growth. It is also indicated as a companion diagnostic to identify patients who are eligible for on-label treatment using one of 17 approved biomarker-directed therapies. The test also reports genomic biomarkers, such as microsatellite instability (MSI) and tumor mutational burden (TMD), that can help to inform the best use of immunotherapies.
Importantly, the Centers for Medicare and Medicaid Services (CMS) immediately agreed to cover the FoundationOne CDx test, making it available to Medicare beneficiaries. “Private payers tend to follow CMS’ lead when it comes to coverage of diagnostic testing and biomarker-directed therapies,” says Smith of IQVIA.
This also raises questions about the consistency in the molecular testing results. For instance, several published studies have revealed broad discrepancies when different testing methodologies are used to confirm the same mutation. One recent study of 3,244 patients found that 150 patients were classified for the mutation ALK+ (anaplastic lymphoma kinase) by either fluorescent in situ hybridization (FISH) or immunohistochemistry (IHC) — but only 80 of the patients had the mutation confirmed by both tests. [3] Such variability—between competing testing methods and even within a single test—has implications for all stakeholders, says Smith. And false negatives during diagnostic testing could create missed opportunities — with potentially dire consequences — for patients who may have benefited from the targeted therapy but did not get it because the target biomarker was not accurately identified during testing.
“The complexity of the current testing environment is untenable,” says Feinberg of Cardinal Health. “I suspect regulatory pressures will be significant in the next few years to create standards across the lab industry to make it more manageable.”
To support such efforts, Merck KGaA and its US affiliate EMD Serono recently published its CDx Guide in 2017, as a resource to provide formalized guidance (based on key regulatory requirements from FDA and the European Union) for pharmaceutical and biotechnology companies that are developing and deploying companion diagnostic tests. The Guide uses a structured, question-based approach to foster critical evaluation and creative thinking about the drug- and CDx-development process, and hopes to encourage the development of an industry-harmonized standard, according to the company. [4]
“The ability to generate a full NGS panel to test for all actionable genetic mutations will eventually enable providers to move past the need to call for multiple, individual screening tests to look for just one type of genetic mutations (such as EGFR, BRCA, HER+2) at a time,” says Dracopoli of PGDx. “While some payers may still only want to pay for the screening of a handful of actionable genes that are relevant for a specific drug, if you have to carry out five or six tests at $300 each, it starts to make sense to consider the broader NGS panel to test for all of them.”
“The key for the manufacturer of biomarker-directed drugs or companion diagnostic testing is to provide the supporting evidence that demonstrates the product’s value in the target population compared to alternative therapies. This is particularly important in Europe where it can mean the difference between being covered at a price reflecting the product’s value or not,” says Debbie Warner, Vice President, Commercial Consulting for Kantar Health. Such efforts should include epidemiology studies quantify potential patient populations, supporting the updating of treatment guidelines, and encouraging favorable reimbursement decisions.
Massive Bio, the company co-founded by Loaiza-Bonilla, for which he serves as Chief Medical Advisor, is helping to advance the field of precision oncology from a patient perspective with its Synergy AI registry, and is collaborating with pharmaceutical companies and healthcare providers to improve education about biomarkers and clinical trial matching. This registry combines genomic/biomarker data with artificial intelligence techniques to remove barriers to access and help justify the cost/benefits to diagnostics and therapeutics, with a major emphasis in clinical trial matching and virtual tumor boards.
References
1. https://www.cancercenter.com/ctca-difference/tapur/
2. https://trkcancer.com/?page=eed7f043-cd99-4033-ab9a-038834d351fd
3. Parallel FISH and immunohistochemical studies of ALK status in 3,244 non-small-cell lung cancers reveal major discordances; J Thorac Oncol. 2014 Mar;9(3):295–306; https://www.ncbi.nlm.nih.gov/pubmed/24518086
4. Josef Straub et al., The Precision Medicine Initative at [German] Merck: State-of-the-Art Patient Selection; The Journal of Precision Medicine, 2017.; https://www.thejournalofprecisionmedicine.com/wp-content/uploads/2017/09/ARTICLE-9.pdf

















